
Primer Design and Phylogenetic Analysis of Codium fragile using the tufA gene
Brief Overview
This post documents a bioinformatics workflow for the algal species Codium fragile, also known colloquially as “Dead Man’s Fingers” due to its finger-like morphology. This workflow demonstrates the process of identification and phylogenetic analysis through the use of the NCBI website, NCBI primer BLAST, and MEGA software with the gene tufA as the target gene. The tufA gene was selected since it is widely considered as a robust barcode for green macroalgae, providing highly distinct interspecific variations and ability to detect cryptic species.
Objectives
- Retrieve tufA sequences of Codium fragile and related algal species from NCBI.
- Align the sequences using ClustalW in MEGA.
- Design primers for amplification of a region using the tufA gene.
- Verify the primer pair using Primer-BLAST.
- Construct a phylogenetic tree using MEGA.
- Analyze the relationships between Codium fragile and related green macroalgae taxa.
Target Organism and Gene
- Target Organism: Codium fragile
- Gene: tufA
- Database: NCBI Nucleotide
- Analysis tools: NCBI, Primer3, Primer-BLAST, MEGA
Sequence Collection from NCBI
The NCBI Nucleotide database was used to retrieve tufA sequences. The target sequence was Codium fragile MK930523.1 and additional tufA sequences from related green algae taxa were downloaded for comparison and phylogenetic analysis.
Table 1: Sequence information for the tufA gene in the target species (Codium fragile) and four additional related taxa.
| Species | Gene | Access Number | Sequence type | Database |
|---|---|---|---|---|
| Codium fragile | tufA | MK930523.1 | partial cds; chloroplast | NCBI Nucleotide |
| Codium hubbsii | tufA | KP685854.1 | partial cds | NCBI Nucleotide |
| Codium minus | tufA | KP685853.1 | partial cds | NCBI Nucleotide |
| Caulerpa taxifolia | tufA | PP313095.1 | partial cds; chloroplast | NCBI Nucleotide |
| Halimeda cf. cuneata | tufA | FJ624538.1 | partial cds; chloroplast | NCBI Nucleotide |

Figure 1: FASTA sequence for the tufA gene of the target species Codium fragile.
Alignment of Sequences
All five tufA sequences were imported into MEGA and aligned using ClustalW. The sequence type was DNA. Regions of variation were observed, including single nucleotide changes (SNPs) and indels.
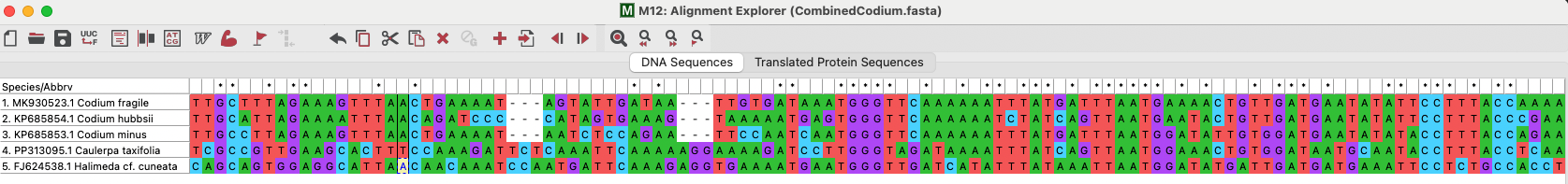
Figure 2: Screenshot of a section of the sequence alignment. Note the dashes indicating indels, and the color differences in the columns indicating SNPs.
Primer Design
Due to tufA being a protein encoding gene, third-position wobble SNPs were observed throughout the alignment which made it difficult to identify completely conserved regions of a suitable length (>20 bp) for primer design. As such, I utilized a mostly conserved area to develop an optimized primer pair specific to Codium fragile.
To design specific primers for Codium fragile using the tufA gene, the multiple sequence alignment was reviewed to identify mostly conserved regions. Coordinate boundaries (bases 50-130 and 480-580) were selected using MEGA and input to NCBI Primer-BLAST. The resulting primer pair optimizes melting temperatures (~59°C) and GC content (~50%) while yielding a 518 bp amplicon suitable for down-stream sequencing and identification.
Primers were verified to be specific by using NCBI primer-BLAST with Chlorophyta as the reference organism within the Nucleotide collection (nt) database. The only hits from similar organisms were from Codium fragile, which indicates that there isn’t an issue with primer specificity.

Figure 3: Relevant information about the selected forward and reverse primers.
Table 2: Primer information and logic for the tufA sequence of Codium fragile (MK930523.1).
| Metric | Forward Primer | Reverse Primer | Logic |
|---|---|---|---|
| Sequence (5’->3’) | GCTGATCAAGTGGATGATGACG | TGGGGTAGAATGGATGATGGT | Clean sequence of a suitable length. |
| Length | 22 bp | 21 bp | Right in the ideal 18-22 bp sweet spot for specificity. |
| Position | Bases 58 to 79 | Bases 555 to 575 | Fits within the mostly conserved area of the sequence. |
| Melting Temp (Tm) | 59.46°C | 58.21°C | They are nearly identical (1.25 degree difference), meaning they will anneal well at the same temperature in a PCR machine. |
| GC Content | 50.00% | 47.62% | Within the ideal 40-60% range, ensuring strong binding without being too sticky. |
| Expected Amplicon Size | 518 bp | This length is short enough to amplify easily and long enough to provide robust phylogenetic resolution. |
Phylogenetic Tree Construction
The software MEGA was used to create a phylogenetic tree based on the aligned tufA sequences of the five taxa. The tree was generated with the neighbor-joining method, the kimura 2-parameter model, and bootstrap analysis with 1000 replications.
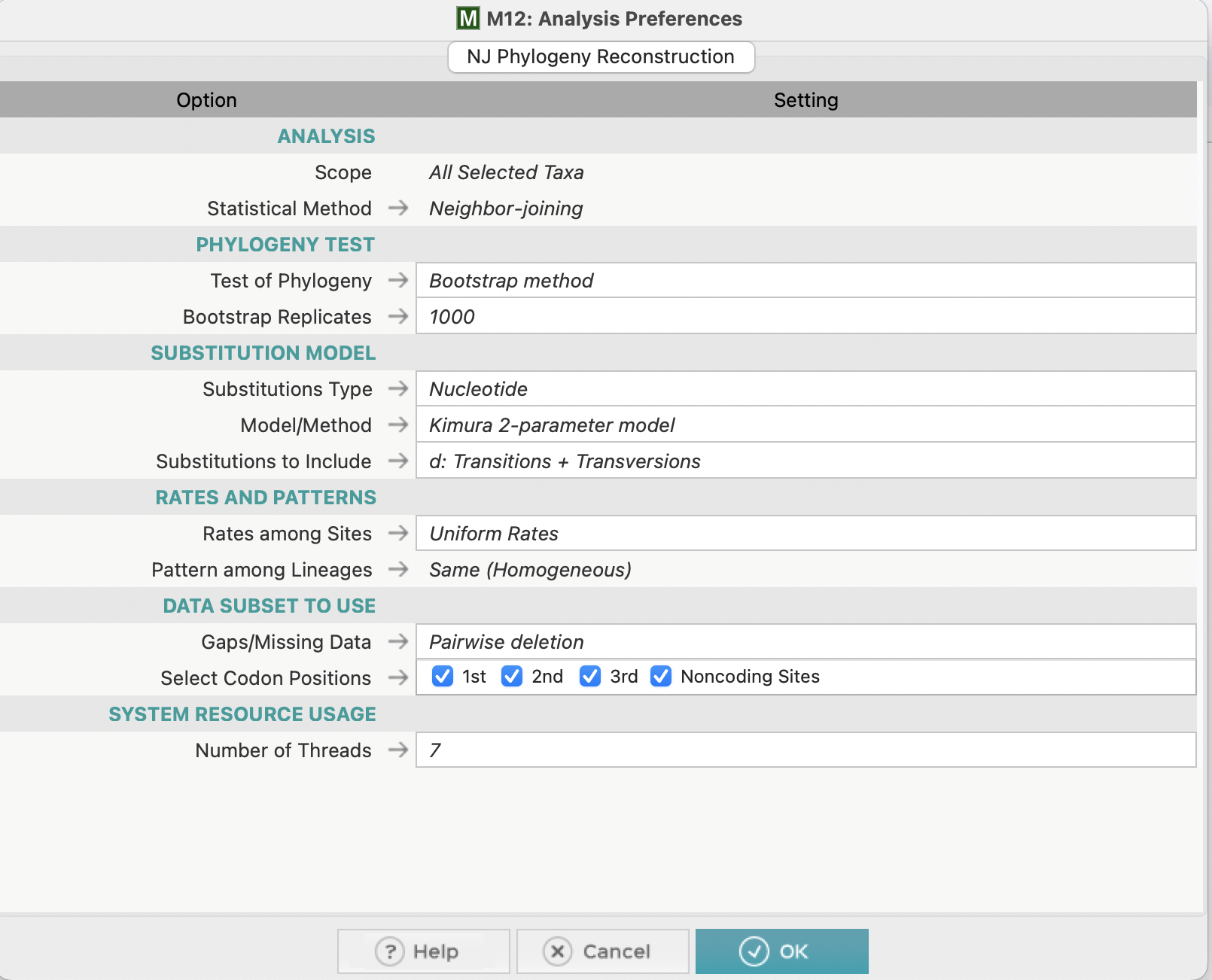
Figure 4: Phylogenetic tree construction parameters screenshot in MEGA software.
Phylogenetic Tree Result and Analysis
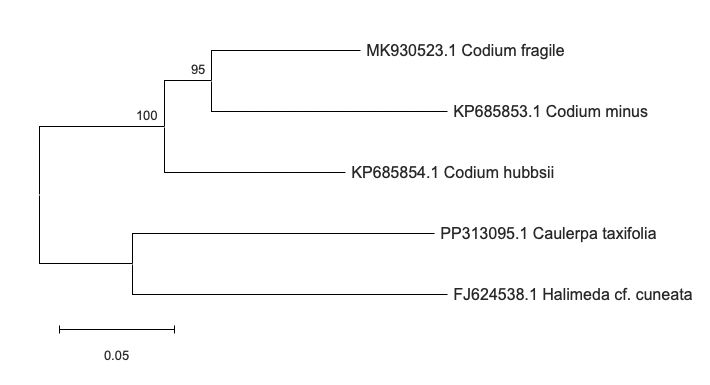
Figure 5: Phylogenetic tree of the target organism (Codium fragile) and four additional taxa based on the tufA gene with 1000 bootstrap runs.
The resulting phylogenetic tree showed a separation between two groups: the Codium genus and the other two green algae species. Codium fragile clustered with both Codium minus and Codium hubbsii with a strong bootstrap value of 100, but Codium fragile clustered even closer to Codium minus than to Codium hubbsii with a bootstrap value of 95. On the other hand, a secondary branch with the other green algae taxa appeared that was separate from the Codium group.
Ultimately, these results indicate that the Codium groups are more closely related to each other than to the other green algae species in the tufA-based analysis. Additionally, Codium fragile and Codium minus seem to be more closely related to each other than they are to Codium hubbsii.
Conclusion
This bioinformatics workflow demonstrated the process of phylogenetic analysis for green algae species. The tufA gene sequences of Codium fragile was used to design primers with NCBI primer BLAST, were verified for specificity, and the primer pair produced a 518 bp amplicon with the necessary parameters for PCR analysis (Tm, GC content, primer length). MEGA software was used to align the sequences of the five taxa and construct a phylogenetic tree based on the tufA sequences. The tree showed that the Codium genus grouped together, while the other taxa were genetically separated. This workflow demonstrates the ability to use the tufA gene for primer design and phylogenetic construction in green algae species.